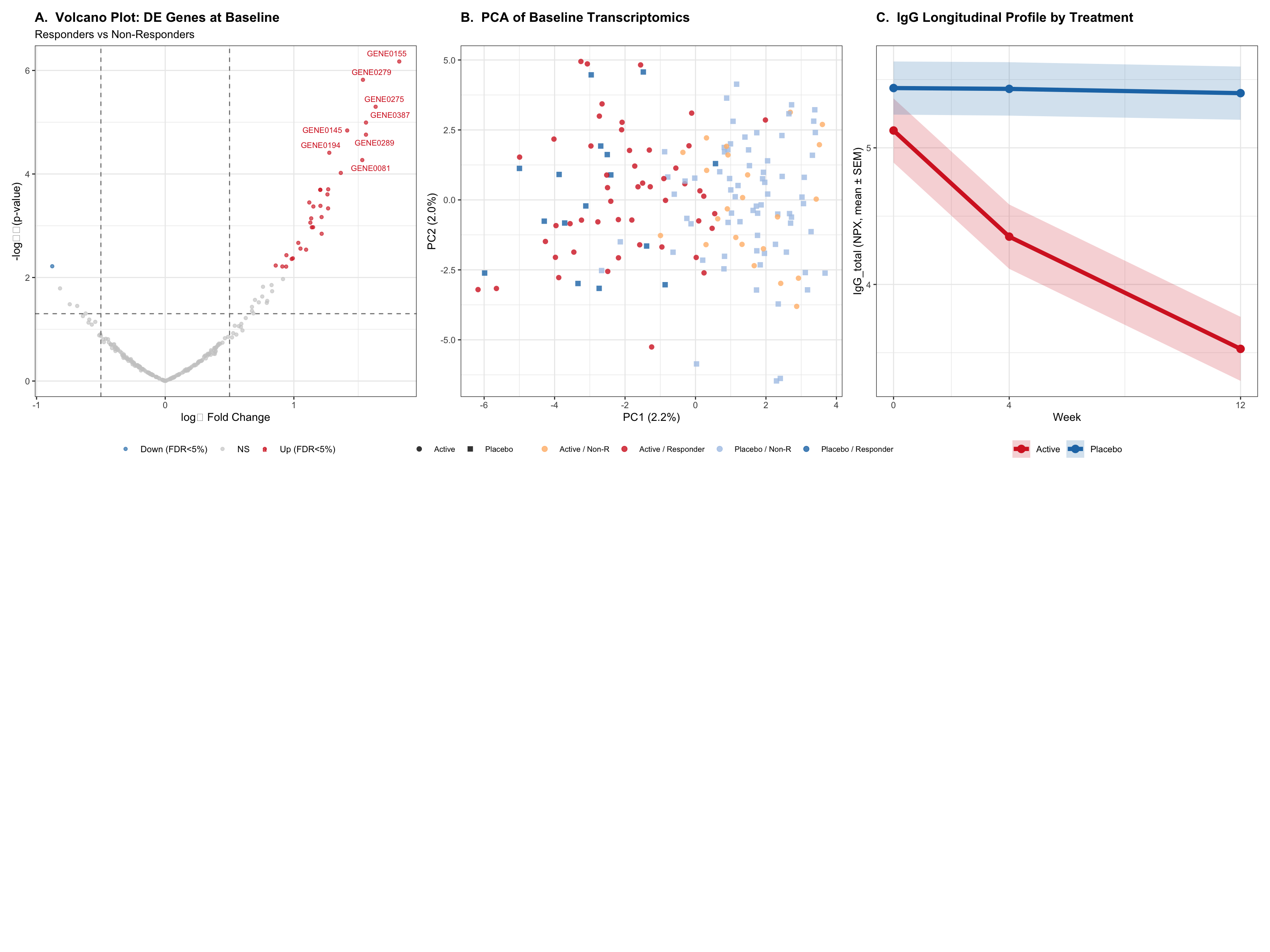
This report analyses simulated Phase II trial data for a PD-1 checkpoint inhibitor (nivolumab-like) in advanced NSCLC. Immune checkpoint blockade (ICB) reinvigorates exhausted tumour-infiltrating T cells, restoring anti-tumour immunity. Response is heterogeneous and biomarker-driven — making this an ideal setting for multi-omics biomarker discovery. Three analytical streams are applied:
Multi-omics — tumour transcriptomics (immune gene signatures) and Olink immune proteomics (primary readout: tumour mutational burden proxy via TMB)
ML pipeline — UMAP of immune phenotypes, k-means patient stratification, elastic-net + random forest ORR prediction
1 Background & Objectives
1.1 Scientific Rationale
PD-1 (programmed cell death protein 1) is an inhibitory receptor expressed on activated T cells. Tumour cells exploit the PD-1/PD-L1 axis to evade immune surveillance. Anti-PD-1 monoclonal antibodies (nivolumab, pembrolizumab) block this interaction, restoring cytotoxic T-lymphocyte activity against tumour antigens.
Key predictive biomarkers in NSCLC:
Biomarker
Platform
Clinical use
PD-L1 IHC (TPS/CPS)
Immunohistochemistry
Pembrolizumab eligibility threshold
TMB (tumour mutational burden)
Whole exome / panel sequencing
Higher TMB → more neoantigens → higher ORR
MSI/dMMR
IHC / PCR
Pan-cancer pembrolizumab approval
ctDNA dynamics
Liquid biopsy
Early response / resistance monitoring
CD8+ TIL density
IHC / deconvolution
Inflamed vs excluded vs desert phenotype
Tumour immune microenvironment (TIME) phenotypes:
Inflamed (T-cell rich): High CD8, PD-L1+, responsive to ICB
Immune excluded: T cells at tumour margins; stromal barriers (TGF-β, VEGF)
Immune desert: Low TIL density; primary resistance; requires combination strategies
1.2 Study Objectives
Identify baseline transcriptomic immune signatures (inflamed vs excluded vs desert) associated with objective response
Characterise TMB and Olink cytokine/immune protein dynamics during ICB treatment
Quantify progression-free survival (PFS) differences between biomarker-high and biomarker-low patients
Build a multi-feature ML classifier combining TMB, PD-L1 proxies, and immune gene expression for ORR prediction
All data are fully synthetic (seed = 123). The simulation encodes realistic biological structure: batch effects in transcriptomics, Emax-shaped primary biomarker trajectories, and gene-expression-linked responder status calibrated to the Non-Small Cell Lung Cancer (NSCLC) setting.