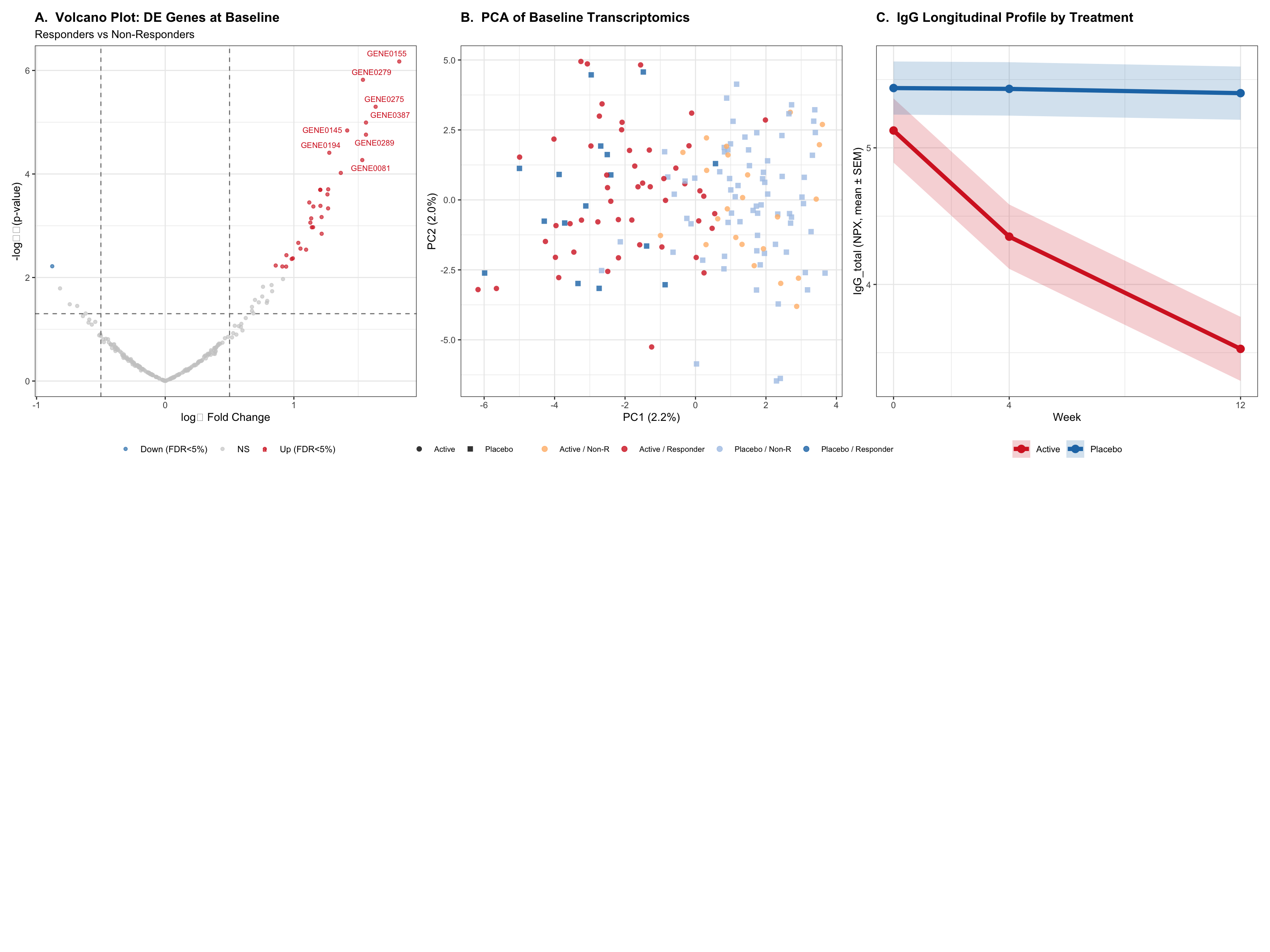
This report analyses simulated Phase II trial data for a PCSK9 inhibitor (evolocumab-like) in heterozygous familial hypercholesterolaemia (HeFH). PCSK9 inhibition upregulates hepatic LDL receptors, markedly reducing LDL-C — the primary causal risk factor for atherosclerotic cardiovascular disease (ASCVD). Three analytical streams are applied:
ML pipeline — patient phenotyping, UMAP of lipid/inflammatory profiles, elastic-net + random forest LDL-C response prediction
1 Background & Objectives
1.1 Scientific Rationale
PCSK9 (proprotein convertase subtilisin/kexin type 9) is a serine protease secreted by hepatocytes that binds the LDL receptor (LDLR) and directs it towards lysosomal degradation. This reduces LDLR recycling to the cell surface, impairing LDL-C clearance. Anti-PCSK9 monoclonal antibodies (evolocumab, alirocumab) prevent PCSK9–LDLR binding, increasing LDLR density and dramatically lowering circulating LDL-C.
Lipid biomarker hierarchy in ASCVD risk:
Biomarker
Role
PCSK9i effect
LDL-C (primary)
Causal ASCVD driver
−50 to −70% from baseline
ApoB
LDL particle number (more predictive)
−40 to −55%
Lp(a)
Genetic residual risk; modest PCSK9i effect
−20 to −30%
hsCRP
Inflammatory risk (statin-responsive)
Minimal direct effect
Non-HDL-C
Includes all atherogenic particles
−50 to −60%
HeFH clinical context:
Prevalence: ~1:250 globally; severely elevated LDL-C from birth
Statin intolerance / inadequate LDL-C control common in HeFH
PCSK9 inhibitors achieve guideline LDL-C targets (<1.4 mmol/L) in >70% of HeFH patients
FOURIER (evolocumab) and ODYSSEY OUTCOMES (alirocumab) demonstrated significant MACE reduction
1.2 Study Objectives
Characterise the transcriptomic signature of LDL-C super-responders vs partial responders at week 12
Quantify LDL-C, ApoB, and Lp(a) NPX trajectories using an Emax PK/PD framework
Assess time-to-LDL-target (< guideline threshold) and MACE-surrogate endpoint differences between responder strata
Develop a baseline multi-omics classifier for LDL-C response prediction to guide patient selection
All data are fully synthetic (seed = 456). The simulation encodes realistic biological structure: batch effects in transcriptomics, Emax-shaped primary biomarker trajectories, and gene-expression-linked responder status calibrated to the Heterozygous Familial Hypercholesterolaemia (HeFH) setting.