Neurology · Anti-Amyloid mAb (lecanemab-like) · Early Alzheimer’s Disease
Executive Summary
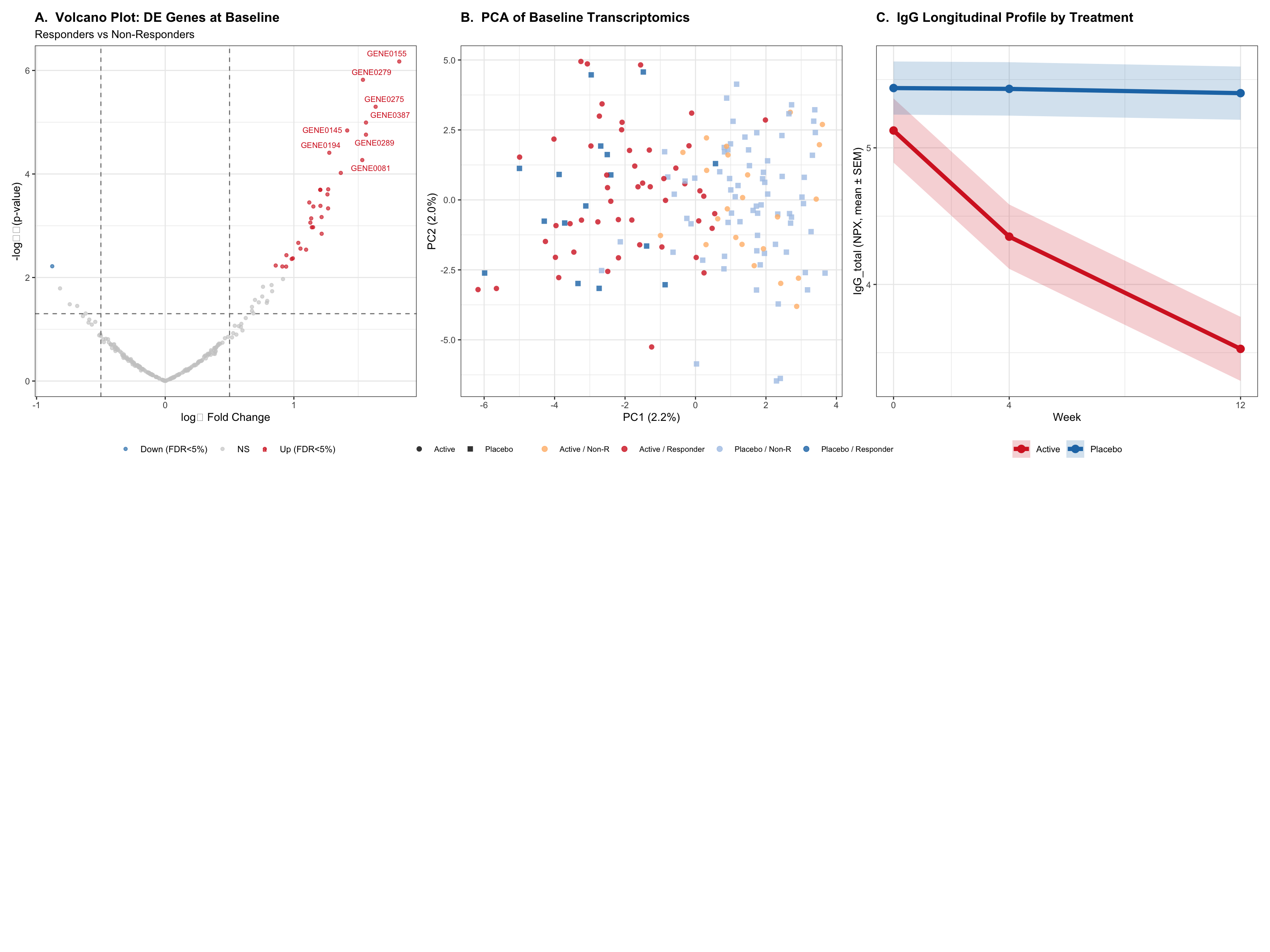
This report analyses simulated Phase III trial data for an anti-amyloid monoclonal antibody (lecanemab-like / LEQEMBI) in early Alzheimer’s disease. Lecanemab targets soluble amyloid-β protofibrils — the toxic intermediate species upstream of plaque formation. The analysis integrates:
Multi-omics — neuroinflammatory transcriptomics and Olink neurology panel proteomics (primary readout: plasma p-tau181 NPX, a fluid biomarker of AD pathology)
ML pipeline — patient stratification by biomarker profile, UMAP of CSF/plasma proteome, elastic-net + random forest prediction of 18-month clinical responders
1 Background & Objectives
1.1 Scientific Rationale
The amyloid cascade hypothesis posits that accumulation of amyloid-β (Aβ) — initially as soluble oligomers and protofibrils, then as insoluble plaques — triggers downstream tau hyperphosphorylation, neurofibrillary tangle formation, neuroinflammation, synaptic loss, and ultimately neurodegeneration. Lecanemab (BAN2401) preferentially binds soluble Aβ protofibrils, removing toxic species before plaque consolidation.
Fluid biomarker landscape in Alzheimer’s disease:
Biomarker
Matrix
Biological meaning
Direction in AD
Aβ42/40 ratio
CSF / plasma
Amyloid plaque burden
↓ (sequestered in plaques)
p-tau181 / p-tau217
CSF / plasma
Tau phosphorylation; AD-specific
↑
Total tau (t-tau)
CSF
Neurodegeneration / axonal damage
↑
NfL (neurofilament light)
CSF / plasma
Non-specific neurodegeneration
↑
GFAP (glial fibrillary acidic protein)
CSF / plasma
Astrocyte activation
↑
Key trial design elements:
Population: Amyloid-positive (PET or CSF) early AD (MCI or mild dementia; CDR 0.5–1)
Primary endpoint: CDR-SB change from baseline at 18 months
Key secondary: Amyloid PET centiloid change; Aβ42/40 ratio normalisation
Safety: ARIA-E and ARIA-H monitoring (amyloid-related imaging abnormalities)
LEQEMBI CLARITY AD (Phase III): 27% slowing of CDR-SB decline vs placebo
1.2 Study Objectives
Identify baseline plasma proteomic profiles (p-tau181, Aβ42/40, NfL, GFAP) that predict cognitive response at 18 months
Quantify p-tau181 NPX dynamics under anti-amyloid therapy using an Emax amyloid-clearing PD framework
All data are fully synthetic (seed = 789). The simulation encodes realistic biological structure: batch effects in transcriptomics, Emax-shaped primary biomarker trajectories, and gene-expression-linked responder status calibrated to the Early Alzheimer’s Disease (MCI / mild dementia) setting.