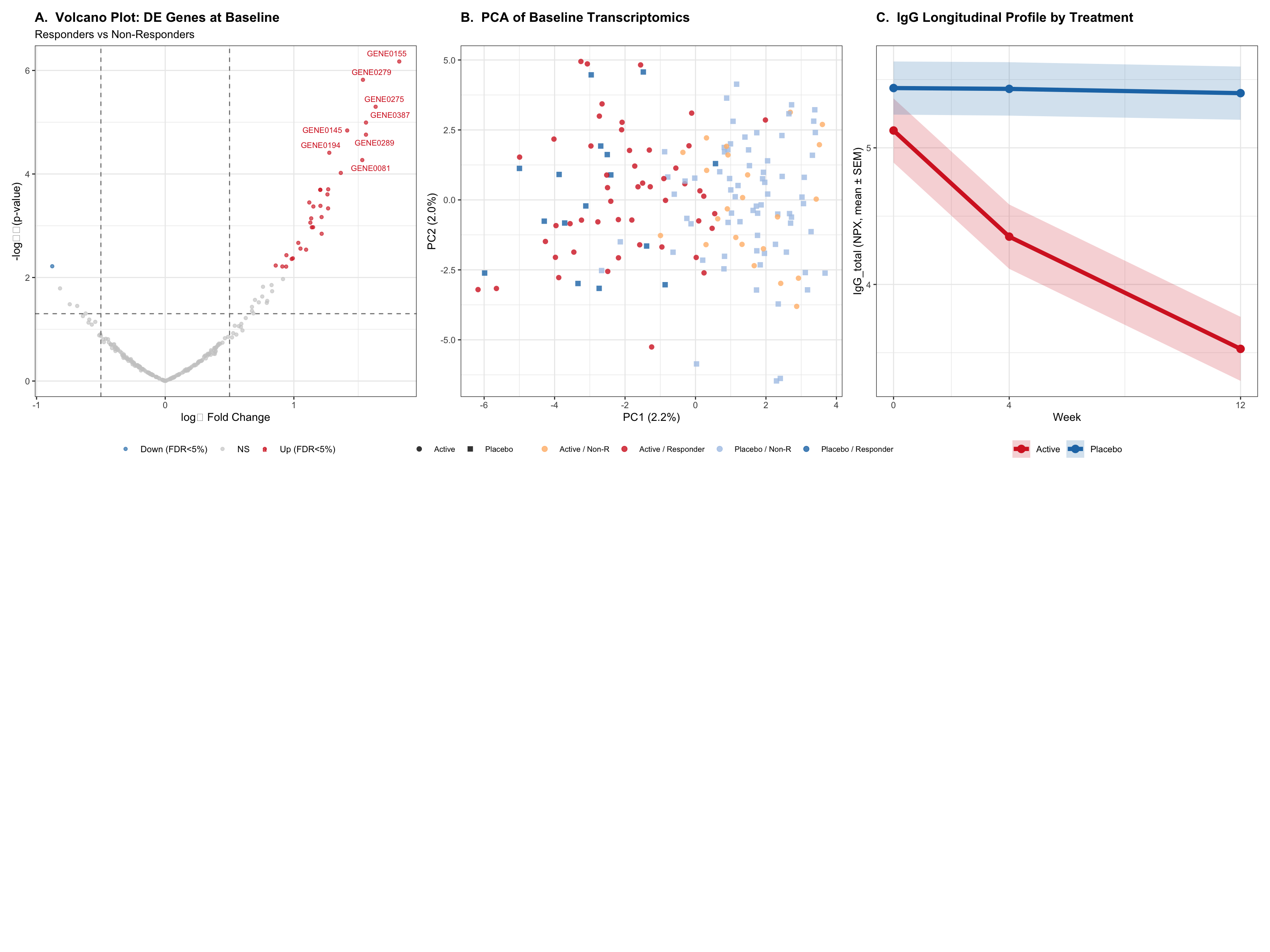
This report analyses simulated Phase II trial data for an FcRn inhibitor (efgartigimod alfa / VYVGART-like) in generalised Myasthenia Gravis (gMG), an IgG-mediated autoimmune neuromuscular disorder. The analysis integrates three analytical streams:
Multi-omics — transcriptomic differential expression and Olink NPX proteomics (primary readout: IgG total NPX)
ML pipeline — UMAP, k-means clustering, elastic-net + random forest response prediction
1 Background & Objectives
1.1 Scientific Rationale
The neonatal Fc receptor (FcRn) salvages IgG antibodies from lysosomal degradation, recycling them into circulation. In gMG, pathogenic anti-AChR and anti-MuSK IgG antibodies impair neuromuscular junction transmission. FcRn inhibition with efgartigimod accelerates IgG catabolism, reducing all IgG subclasses including pathogenic autoantibodies — a mechanism validated across gMG, ITP, CIDP, and pemphigus vulgaris.
All data are fully synthetic (seed = 237). The simulation encodes realistic biological structure: batch effects in transcriptomics, Emax-shaped primary biomarker trajectories, and gene-expression-linked responder status calibrated to the Generalised Myasthenia Gravis (gMG) setting.